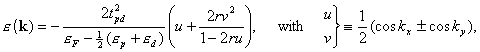Activities 1995-2000
Alkali-doped Fullerenes
Clusters
Cuprates
GW Method
High-Temperature Superconductors
LMTO-based methods
LmtART
Linear Response DF-LMTO method
Quantum Monte-Carlo
Stuttgart TB-LMTO-ASA Program
Third-generation MTO
A substantial effort, centered around Olle Gunnarsson, has gone into work on the alkali-doped fullerenes. These systems are novel in the sense that the energy scales for electrons and phonons are comparable, and this leads to interesting issues. There are many competing effects, which make the physics rich and pose a challenge to the theoretical treatment and, as we have demonstrated, many properties can be calculated thanks to the clear separation of these solids into weakly-coupled C60-molecules. This causes the above-mentioned common energy range for the electronic bandwidths and molecular vibrations to be distinct from that of the intra-molecular electronic energies, as well as from that of the inter-molecular phononic energies. The parameters of the basic Hamiltonian for the electron-phonon system was obtained in the early nineties using DF calculations and subsequent adjustments to experiments. The electronic band structures are so simple that they can be given analytically [1].
One important problem is the superconductivity. It is determined both
by the electron-phonon coupling, ![]() ,
and the Coulomb pseudopotential,
,
and the Coulomb pseudopotential,![]() ,
which describes the effects of the Coulomb repulsion. In cooperation with
the experimental group of W. Eberhardt in Jülich, the electron-phonon
coupling parameters were extracted from photoemission experiments for free
C60 molecules and it was found that
,
which describes the effects of the Coulomb repulsion. In cooperation with
the experimental group of W. Eberhardt in Jülich, the electron-phonon
coupling parameters were extracted from photoemission experiments for free
C60 molecules and it was found that ![]() [2].
For conventional superconductors, retardation effects are believed to reduce
[2].
For conventional superconductors, retardation effects are believed to reduce![]() to a value of about 0.1, but for the alkali-doped fullerenes, A3C60
(A=Rb or Cs), we found that these effects are relatively small due to the
similarity of the energy scales for phonons and electrons and that, instead,
screening effects reduce
to a value of about 0.1, but for the alkali-doped fullerenes, A3C60
(A=Rb or Cs), we found that these effects are relatively small due to the
similarity of the energy scales for phonons and electrons and that, instead,
screening effects reduce ![]() [3].
As a result,
[3].
As a result, ![]() is substantially larger than for conventional superconductors, specifically
0.3-0.4. This agrees with recent experimental estimates and does not suffice
to suppress the electron-phonon mechanism for superconductivity.
is substantially larger than for conventional superconductors, specifically
0.3-0.4. This agrees with recent experimental estimates and does not suffice
to suppress the electron-phonon mechanism for superconductivity.
Another important issue is why not all alkali-doped fullerenes are insulators, as one might expect from estimates of the Coulomb interaction, U, and the band width, W. We have studied the effects of the orbital degeneracy [4], the band filling, and the lattice structure [5]. We find that the orbital degeneracy substantially increases the ratio U/W for which a Mott transition occurs. The lattice structure is important for the difference between A3C60 (metal) and A4C60 (insulator). We also find that the electron-phonon interaction plays a substantial role [5], with an interesting competition between the Jahn-Teller effect and the Hund's rule coupling.
The electrical resistivity of metallic A3C60 is exceptionally large, corresponding to an apparent mean free path l ~ 1-2 Å at large temperatures T. Thus l is much shorter than the separation d =10 Å between two molecules. This contradicts an observation made for many systems, i.e. that the resistivity saturates when l ~ d, earlier considered as a universal behavior. We have performed essentially exact quantum Monte-Carlo calculations for a model of A3C60 , including the scattering against intra-molecular phonons. We find that for large T indeed l <<d . This illustrates that saturation when l ~ d is not a universal phenomenon [6]. The reasons for the lack of saturation were analyzed, and it was found that the small electronic bandwidth and the coupling to intra-molecular phonons are important [See also: FKF Annual Report 1999, p I-100-104].
A review of superconductivity in the fullerenes has been written [7].
The alkali-doped fullerenes raise some fundamental questions, e.g., whether we can have l <<d for the resistivity, which can be answered only by accurate many-body calculations without uncontrolled approximations. We have therefore developed the tools for doing quantum Monte-Carlo calculations of ground-state properties at T = 0 [4] and dynamical correlation functions for T > 0 [6]. The first method uses a projection method approach and a fixed node approximation, while the second method uses a determinantal method. The T > 0 method gives essentially exact results for the electron-phonon problem to which it was applied above, and we have demonstrated that the fixed-node approximation is rather accurate for the problems where it was used.
The so-called sudden approximation is applied in almost all calculations of photo-emission. We have studied its validity for the case of coupling to localized excitations. We found that the sudden approximation becomes valid for rather small photon energies, in contrast to the case of coupling to plasmons. The relevant energy scales for the validity of the sudden approximation were determined [8].
Experimentally, the core level photoemission spectra of cuprates have been found to show a clear dependence on the Cu-O network, in spite of the locality of the probe. We have calculated the spectrum in the Anderson impurity model, determining the parameters from ab initio DF-LMTO calculations [9]. The model was solved using an expansion in the inverse degeneracy, developed in the group. Fig. 1 illustrates the good agreement with experiment. The leading peak results from a valence electron filling the Cu 3d level to screen the Cu core hole in the photoemission process. The core spectrum therefore depends on the valence electronic structure. In Bi2CuO4, Ba3Cu2O4Cl2and Li2CuO2 there is little hopping between the Cu atoms because they belong to strings of edge-sharing, and therefore uncoupled CuO4-squares. As a result, the screening electron comes from a narrow resonance, leading to a narrow peak. In Sr2CuO3 and SrCuO2, the CuO4-squares form corner-sharing -and therefore strongly coupled- 1D networks. In Sr2CuO2Cl2the CuO4-squares form corner-sharing 2D networks, like in the superconducting cuprates, and in Ba2Cu3O4Cl2 with extra, weakly coupled Cu atoms at the center of every second O4-square. LaCuO3 has a 3D Cu-O network. In all these systems additional screening channels enter, leading to more structured leading peaks.
Contrary to the case for the fullerenes, the mechanism behind the superconductivity in the cuprates remains a major puzzle. These high-temperature superconductors (HTSCs) are doped Mott-Hubbard insulators in which superconductivity occurs below a critical temperature, Tc(p). The latter depends parabolically on the hole-doping p with a maximum at so-called optimal doping. For different materials, maxpTc varies substantially.
Research in our group was initially focussed on DF (LDA) calculations for the optimally doped, stoichiometric YBa2Cu3O7 of energy bands, optical spectra, phonon frequencies in the normal and superconducting phases, the electron-phonon interaction, Raman spectra, and the Fermi surface. The LDA, however, completely fails to describe the electronic structure of the undoped, insulating compounds, and this lead Zaanen et al. to develop the LDA+U method for magnetically ordered Mott-Hubbard insulators at the beginning of the nineties. In this method, the LDA is corrected by addition of the on-site Coulomb interactions, U for the correlated electrons in a static mean-field treatment. Using the LDA+U, Lichtenstein et al. now argued that orbital polarization should be treated on equal footing with spin polarization, and they demonstrated that orbital ordering is a necessary condition for obtaining the crystal structure and exchange couplings observed for the 3d Mott-Hubbard insulator KCuF3[10]. This early work fertilized research on orbital polarization in transition-metal compounds. Another application of the LDA+U formalism, this time to the 4f-electrons in REBa2Cu3O7 , enabled Lichtenstein and Mazin to explain why Pr-doping suppresses superconductivity more efficiently in NdBa2Cu3O7 than in YBa2Cu3O7[11]. Most recently, Dudarev et al. compared the charge density in NiO measured by high-energy electron diffraction with those calculated using various DF approximations. The best match to the experimental charge density was obtained by the generalized gradient approximation (GGA) and the LDA+U with U and J adjusted, but neither method sufficently suppresses the covalent charge density between Ni and O [12].
Whereas the LDA (+U ) gives a parameter-free and reasonable description
of the high-energy properties of the HTSCs and produces all electronic
bands in a broad energy range, almost any theory of the low-energy
properties uses a one-band, correlated model Hamiltonian. The rationale
for this is that, since the superconductivity originates in the CuO2
-layers and a single layer gives rise to merely one band at the Fermi level,
a one-band model should suffice. We have developed a generally applicable
method for computing, from a given single-electron potential, the Wannier-like
orbitals
which
form a basis for such low-energy, few-band Hamiltonians [13(a),13(b)].
The LDA result for a HTSC is shown in Fig. 2.
This orbital is centered on Cu, has dx2-y2symmetry,
and extends to the 2nd-nearest neighbor oxygen atoms. As a consequence,
its integrals for single-particle hopping on the Cu-centered square lattice,
t,
t', t'', ..., have rather long range. We have succeeded
in computing this orbital and its hopping integrals for a series
of HTSC materials with single layers and find that there is essentially
only one parameter which depends on the material, namely the range-parameter ![]() ,
which
gives the ratio, t(n)/t,
of
any hopping integral to the nearest-neighbor one. Most importantly, our
calculated r-values shown in Fig. 3 correlate
with the observed max Tc. To understand why, is a key
to understanding the mechanism of HTSC and poses a challenge to many-body
theorists, such as those who currently use a k-space renormalization-group
method to search for the leading correlation-driven instabilities for various
band shapes.
,
which
gives the ratio, t(n)/t,
of
any hopping integral to the nearest-neighbor one. Most importantly, our
calculated r-values shown in Fig. 3 correlate
with the observed max Tc. To understand why, is a key
to understanding the mechanism of HTSC and poses a challenge to many-body
theorists, such as those who currently use a k-space renormalization-group
method to search for the leading correlation-driven instabilities for various
band shapes.
We have understood the much simpler issue of how r is controlled by structural and chemical factors, such as the distance between copper and apical oxygen [14, 15]. In order to explain this, we must first transform to a larger basis of orbitals which are more localized than the one shown on the front page. It is customary to use Ox px , Cu dx2-y2 , and Oy py, in which case the nearest-neighbor Cu-Cu hopping (t) is described as: Cu dx2-y2 Oy py Cu dx2-y2 , while the 2nd and farther Cu-Cu hoppings require various intermediate O-O hoppings. We have found that by adding one further -diffuse Cu 4s-like- orbital with axial symmetry, the resulting four orbitals are so localized that merely nearest-neighbor hoppings (Cu and O) need to be included. Hence, the various O-O hoppings of the 3-orbital model, and thereby t', t'', ..., of the 1-orbital model, all proceed via the axial orbital, and this leads to relations like: t' = 2t''. In the 4-orbital model, the dispersion of the bare conduction band is simply:
|
(1) |
and the range-parameter is essentially the Cu s character:
|
(2) |
The energy ![]() of the axial orbital is eV's above the Fermi level,
of the axial orbital is eV's above the Fermi level, ![]() ,
which itself is eV's above the O p-level,
,
which itself is eV's above the O p-level, ![]() .
Expansion of (1-2ru)-1
yields the one-band form:
.
Expansion of (1-2ru)-1
yields the one-band form:
The axial orbital not only provides the dominant channel for the intra-layer
hopping beyond nearest Cu-neighbors, but also provides the dominant channel
for single-particle hopping out of the layer, ![]() ,
which according to Eq. (1) depends on k like
v2
which
has
,
which according to Eq. (1) depends on k like
v2
which
has ![]() as
a nodal line [14]. For
materials like La2CuO4,
Bi2Sr2CuO4,
and Tl2Ba2CuO6,
where the stacking of the CuO2-layers is
body-centered, the inter-layer hopping has an additional nodal line along
as
a nodal line [14]. For
materials like La2CuO4,
Bi2Sr2CuO4,
and Tl2Ba2CuO6,
where the stacking of the CuO2-layers is
body-centered, the inter-layer hopping has an additional nodal line along ![]() .
This supresses the perpendicular transport significantly, and provides
a strong argument against the so-called inter-layer tunnelling mechanism
for HTSC [15].
.
This supresses the perpendicular transport significantly, and provides
a strong argument against the so-called inter-layer tunnelling mechanism
for HTSC [15].
In multi-layer HTSCs, the neighboring CuO2-layers
are stacked on top of each other and the integral for hopping between the
layers proceeds mainly from Cu s to Cu s[14].
Our predicted value of the inter-layer exchange coupling in the bi-layer
insulator YBa2Cu3O6
was verified by Keimer et al. using neutron scattering. Also for metallic
multi-layer HTSCs, it is usually believed that the Coulomb repulsion localizes
the electrons onto the individual layers. If that is not the case, there
will be electronic subbands whose (bare) dispersions are given by Eqs.(1)-(2),
but with ![]() added to
added to ![]() for a bi-layer,
for a bi-layer, ![]() and 0 for a triple-layer, and
and 0 for a triple-layer, and![]() for an infinite layer. Hence, the subbands will have different r-values
and the lowest, most bonding subband will have the highest r. For
bi-layer YBa2Cu3O7
with maxTc we find
for an infinite layer. Hence, the subbands will have different r-values
and the lowest, most bonding subband will have the highest r. For
bi-layer YBa2Cu3O7
with maxTc we find ![]() ,
for triple-layer HgBa2Ca2Cu3O4
with maxTc=135K we find
,
for triple-layer HgBa2Ca2Cu3O4
with maxTc=135K we find ![]() and 0.37, and for infinite-layer CaCuO2 we find r ranging
from 28 to 0.42. We thus have the unexpected result that the correlation
between r and maxTc
observed for single-layer HTSCs, extends to multi-layer HTSCs with up to
3 layers, provided that most bonding subband is considered! This speaks
against the van-Hove scenario, because for YBa2Cu3O7
it is the saddle-point of the anti-bonding band which is close to
and 0.37, and for infinite-layer CaCuO2 we find r ranging
from 28 to 0.42. We thus have the unexpected result that the correlation
between r and maxTc
observed for single-layer HTSCs, extends to multi-layer HTSCs with up to
3 layers, provided that most bonding subband is considered! This speaks
against the van-Hove scenario, because for YBa2Cu3O7
it is the saddle-point of the anti-bonding band which is close to ![]() but it is consistent with our finding that for YBa2Cu3O6+y
it is the bonding subband which can support anti-ferromagnetic spin-fluctuations [16].
but it is consistent with our finding that for YBa2Cu3O6+y
it is the bonding subband which can support anti-ferromagnetic spin-fluctuations [16].
In bi-layer YBa2Cu3O7
the oxygens are dimpled out of the Cu-plane by ![]() towards Y and away from Ba. This allows for a linear electron-phonon coupling
of the corresponding modes, of which the Raman-active ones were studied
intensively in Cardona's and our groups at the beginning of the nineties.
We found these modes to have relatively strong electron-phonon couplings,
despite the smallness of the static dimple. The reason is that when the
energy
towards Y and away from Ba. This allows for a linear electron-phonon coupling
of the corresponding modes, of which the Raman-active ones were studied
intensively in Cardona's and our groups at the beginning of the nineties.
We found these modes to have relatively strong electron-phonon couplings,
despite the smallness of the static dimple. The reason is that when the
energy ![]() of the axial orbital is low, it pushes the
of the axial orbital is low, it pushes the ![]() -like
conduction-band orbital down in energy so that it becomes nearly degenerate
with the
-like
conduction-band orbital down in energy so that it becomes nearly degenerate
with the ![]() -orbitals,
whereby the hopping between them may be strong. This near-degeneracy term
adds to the usual change of tpd with Cu-O bond-length
and angle. By adding the Ox
pzand
Oy
pz
orbitals
to the above-mentioned 4-orbital model, the matrix elements for scattering
of an electron from k to
k'
-orbitals,
whereby the hopping between them may be strong. This near-degeneracy term
adds to the usual change of tpd with Cu-O bond-length
and angle. By adding the Ox
pzand
Oy
pz
orbitals
to the above-mentioned 4-orbital model, the matrix elements for scattering
of an electron from k to
k'![]() k+q
by the
k+q
by the ![]() buckling modes of wavevector q can be given analytically [17(a),17(b),17(c)].
For the Ox-Oy mode, this scattering
is maximal when
k=
k'= (p, 0)
and it vanishes when k=(p, 0) and
k'
=(0 ,p). Since the observed
dx2-y2
-wave pairing is presumably caused by a U-driven repulsive
pairing interaction which vanishes at small q and peaks for q
buckling modes of wavevector q can be given analytically [17(a),17(b),17(c)].
For the Ox-Oy mode, this scattering
is maximal when
k=
k'= (p, 0)
and it vanishes when k=(p, 0) and
k'
=(0 ,p). Since the observed
dx2-y2
-wave pairing is presumably caused by a U-driven repulsive
pairing interaction which vanishes at small q and peaks for q ![]() (p, p), this buckling mode provides a pairing
interaction which, although being attractive, might enhance dx2-y2-wave
pairing. The electron-phonon coupling constants for both buckling modes,
(p, p), this buckling mode provides a pairing
interaction which, although being attractive, might enhance dx2-y2-wave
pairing. The electron-phonon coupling constants for both buckling modes, ![]() x²y²
,
turn out to be identical in the model, and equal to a positive number
times the density of states on the Fermi surface weighted by v2.
When the Fermi level is at the van Hove singularity,
x²y²
,
turn out to be identical in the model, and equal to a positive number
times the density of states on the Fermi surface weighted by v2.
When the Fermi level is at the van Hove singularity, ![]() x²y²=
x²y²=![]() s.
This speculation about the buckling modes was confirmed by a full LDA-LMTO
linear-response calculation for hole-doped CaCuO2 of the equilibrium
structure, all phonon branches, and their electron-phonon coupling constants
in both s- and dx2-y2-wave
channels [17(a),17(b),17(c)].
This calculation showed that the mirror symmetry around a CuO2-plane
is broken to develop a static OxOy-dimple, that
s.
This speculation about the buckling modes was confirmed by a full LDA-LMTO
linear-response calculation for hole-doped CaCuO2 of the equilibrium
structure, all phonon branches, and their electron-phonon coupling constants
in both s- and dx2-y2-wave
channels [17(a),17(b),17(c)].
This calculation showed that the mirror symmetry around a CuO2-plane
is broken to develop a static OxOy-dimple, that![]() x²y²
x²y²![]() 0.3 is contributed predominantly by the buckling modes, and that
0.3 is contributed predominantly by the buckling modes, and that ![]()
![]() 0.4.
0.4.
Over ten years ago, Zaanen and Gunnarsson pointed out that in HTSCs
at low doping concentrations, the holes have a tendency to order along
parallel, magnetic domain lines. Such striped phases have recently drawn
a lot of attention. Magnetic stripes with Q= 2p ![]() were observed in La1.6-1/8Nd0.4Sr1/8CuO4
where they are presumably pinned by the non-superconducting LTT phase.
Recent experiments hint that the stripes might be dynamic and are the cause
of the low-energy incommensurate spin excitations observed in La2-xSrxCuO4
throughout the doping range for superconductivity. Whether such dynamical
stripes are responsible for -or decremental to- HTSC remains to be seen.
Fleck et al. treated the 2D Hubbard model with a dynamical mean-field
scheme, using exact diagonalization, and did find stable [10]-oriented
striped phases for hole dopings between 0.05 and 0.2 [18(a),
18(b)]. Also most features of the photoemission spectra could be reproduced
and interpreted with the subband Hamiltonian generated by the real part
of the dynamical mean field. The energy cost for moving a hole perpendicular
to the stripe was calculated as a function of the range-parameter r
and found to be negative for r
were observed in La1.6-1/8Nd0.4Sr1/8CuO4
where they are presumably pinned by the non-superconducting LTT phase.
Recent experiments hint that the stripes might be dynamic and are the cause
of the low-energy incommensurate spin excitations observed in La2-xSrxCuO4
throughout the doping range for superconductivity. Whether such dynamical
stripes are responsible for -or decremental to- HTSC remains to be seen.
Fleck et al. treated the 2D Hubbard model with a dynamical mean-field
scheme, using exact diagonalization, and did find stable [10]-oriented
striped phases for hole dopings between 0.05 and 0.2 [18(a),
18(b)]. Also most features of the photoemission spectra could be reproduced
and interpreted with the subband Hamiltonian generated by the real part
of the dynamical mean field. The energy cost for moving a hole perpendicular
to the stripe was calculated as a function of the range-parameter r
and found to be negative for r ![]() 0.3.
0.3.
In concluding on HTSCs, possible reasons for the observed correlation between maxTc and the hopping range r may be listed: 1) A longer hopping range screens U better, and maybe even overscreens it locally. 2) t' supresses static stripe order. 3) A low energy of the axial orbital increases the tendency towards buckling and, maybe, formation of bipolarons.
During the last 6 years, several new LMTO-based methods were
developed. These methods treat d- and f-electrons as easily
as s- and p-electrons, and may do so with a small basis set,
because the orbitals are constructed from the muffin-tin (MT) approximation
to the potential in the solid. A MT-potential is spherically symmetric
inside non-overlapping spheres surrounding the atoms and constant in between.
In the interstitial region, a linear muffin-tin orbital (LMTO) of the 1st
generation is a spherical harmonics times the appropriate spherical
Hankel or Neumann function, regular at infinity and with a fixed wavenumber
k0
.
This envelope function is continued smoothly inside any sphere as the appropriate
linear combination of spherical harmonics times radial Schrödinger-equation
solutions, ![]() ,
and their first energy derivatives,
,
and their first energy derivatives, ![]() with the energy
with the energy ![]() chosen at the centre of interest. It is intuitively obvious that the way
in which a linear combination of LMTOs reproduces a wave function with
energy
chosen at the centre of interest. It is intuitively obvious that the way
in which a linear combination of LMTOs reproduces a wave function with
energy ![]() of the solid, is by adding to the head of each LMTO that amount of tails,
which makes the total become
of the solid, is by adding to the head of each LMTO that amount of tails,
which makes the total become ![]() . Hence, the LMTO set spans the wave functions of the MT-potential to linear
order in
. Hence, the LMTO set spans the wave functions of the MT-potential to linear
order in ![]() inside the spheres, but merely to 0th order in the interstitial. For close-packed
solids and low energies, one often uses the atomic-spheres approximation
(ASA), which substitutes the MT-spheres with slightly overlapping (space-filling)
atomic spheres and sets k0=0,
so that the envelope functions become simple multipole potentials,
inside the spheres, but merely to 0th order in the interstitial. For close-packed
solids and low energies, one often uses the atomic-spheres approximation
(ASA), which substitutes the MT-spheres with slightly overlapping (space-filling)
atomic spheres and sets k0=0,
so that the envelope functions become simple multipole potentials, ![]() .
In the ASA, the ingredients to the band structure are potential parameters,
obtained from the radial functions at the sphere radii, sR
and a structure matrix, which specifies the spherical-harmonics
expansion around site R' of an LMTO-envelope at site R.
In open structures, also the potentials in the voids are taken to be spherically
symmetric. The long range of the envelopes with k0=0
were gotten rid of in the early eighties by screening with multipoles
on the neighboring sites. This made it possible to generate the structure
matrix in real space and to use tight-binding (TB) techniques in DF calculations,
e.g. to treat imperfect crystals. The two-centre TB-Hamiltonian is simply
the structure matrix pre- and post-multiplied by potential parameters.
The TB-LMTO set is merely a similarity transformation of the bare set.
Another similarity transformation generates an orthonormal set.
Downfolding
is a third feature of this 2nd-generation method: If one wants to
omit, say the Rlm-orbital from a given basis, one must first transform
to a representation in which the tails of the neighboring envelopes, when
expanded around site R have an lm-projection with the proper
radial logarithmic derivative at the sphere,
.
In the ASA, the ingredients to the band structure are potential parameters,
obtained from the radial functions at the sphere radii, sR
and a structure matrix, which specifies the spherical-harmonics
expansion around site R' of an LMTO-envelope at site R.
In open structures, also the potentials in the voids are taken to be spherically
symmetric. The long range of the envelopes with k0=0
were gotten rid of in the early eighties by screening with multipoles
on the neighboring sites. This made it possible to generate the structure
matrix in real space and to use tight-binding (TB) techniques in DF calculations,
e.g. to treat imperfect crystals. The two-centre TB-Hamiltonian is simply
the structure matrix pre- and post-multiplied by potential parameters.
The TB-LMTO set is merely a similarity transformation of the bare set.
Another similarity transformation generates an orthonormal set.
Downfolding
is a third feature of this 2nd-generation method: If one wants to
omit, say the Rlm-orbital from a given basis, one must first transform
to a representation in which the tails of the neighboring envelopes, when
expanded around site R have an lm-projection with the proper
radial logarithmic derivative at the sphere, ![]() .
After this transformation, the Rlm-orbital can be deleted from the
set, which is now complete to 0th order in the deleted channel. Similarity
transformations and downfolding made the 2nd-generation method intelligible,
fast, and useful beyond the realm of close-packed, crystalline transition
metals.
.
After this transformation, the Rlm-orbital can be deleted from the
set, which is now complete to 0th order in the deleted channel. Similarity
transformations and downfolding made the 2nd-generation method intelligible,
fast, and useful beyond the realm of close-packed, crystalline transition
metals.
Our Stuttgart TB-LMTO-ASA program is in use world wide because it is extremely fast, user-friendly, hardware-independent, and free of charge. Input is the crystal structure and output is the charge- and spin-selfconsistent band structure, the partial densities of states, the Fermi surface, plots of the full charge and spin-densities, the total energy, and the partial pressures. In addition, the program delivers tools for analysing the electronic structure and chemical bonding such as: Orbital-projected band structures, COHPs for describing bond strengths, and electron localization functions (ELFs). The division of space into potential-spheres is done automatically [19]. We maintain the programme, give hands-on courses, and follow up with consultations on specific projects (coauthorship is not a condition).
Hedin's GW approximation for calculating the electron self-energy has been widely applied to sp-electron systems and shown to be quite accurate for these weakly correlated systems. In the early nineties, Aryasetiawan and Gunnarsson developed a GW method based on LMTOs. This made it possible to apply the GW approximation to systems with localized states, e.g., transition metal compounds, and to test its applicability for such strongly correlated systems, in particular NiO [20]. Although the method showed deficiencies for some properties of NiO, it gave surprisingly accurate results for the band structure and magnetic moment. A review of the method was written [21].
For computing total-energy differences associated with symmetry-lowering displacements of the atoms, the ASA is too crude. Various full-potential (FP) LMTO methods were developed in the late eighties and early nineties. In these more accurate methods, the LMTOs are defined with respect to non-overlapping potentials, but since the interstitial region is now large, one must use double- or triple-k sets. A further blow up of the basis set is caused by core states which extend beyond the MT-sphere. The charge density in the interstitial is expanded in Hankel functions or plane waves. Similarity transformations and downfolding are not attempted. Hence, the FP-LMTO methods are numerically accurate, but not really fast and intelligible.
Savrasov developed an FP-LMTO linear-response
method with which phonon dispersions and electron-phonon interactions
can be calculated efficiently throughout the Brillouin zone [22].
Compared with the supercell method used previously for YBa2Cu3O7
this was a big step forward, although for reasons of computer storage only
the simplest HTSC, idealized CaCuO2 could be treated [17(a),17(b),17(c)].
Like the methods of Zein and Baroni, based on plane-waves and pseudopotentials,
Savrasov's method uses the Sternheimer approach and thereby avoids inverting
a large dielectric matrix. To prove the method, Savrasov et al. computed
the phonon dispersions and the electron-phonon spectral functions ![]() for elemental Al, Pb, V, Nb, Ta, Mo, Cu, and Pd, and compared with experimental
tunnelling data, temperature-dependent electrical and thermal resistivities,
as well as the electronic specific heats. Only for Pd did their specific-heat
enhancements -correctly- leave room for a contribution from spin-fluctuations.
It was concluded that, for the materials considered, the electron-phonon
interaction is described with 10 per cent accuracy [22(a),22(b)].
for elemental Al, Pb, V, Nb, Ta, Mo, Cu, and Pd, and compared with experimental
tunnelling data, temperature-dependent electrical and thermal resistivities,
as well as the electronic specific heats. Only for Pd did their specific-heat
enhancements -correctly- leave room for a contribution from spin-fluctuations.
It was concluded that, for the materials considered, the electron-phonon
interaction is described with 10 per cent accuracy [22(a),22(b)].
Next, Savrasov generalized the method to treat spin-fluctuations.
Lacking an accurate approximation for the kernel ![]() describing dynamical exchange-correlation effects in time-dependent DF
theory, the so-called adiabatic GGA was adopted. The efficiency of the
approach was demonstrated by applications to the transverse spin fluctuations
at T=0 in ferromagnetic Fe and Ni, as well as to the paramagnetic
response in Cr and Pd. Some discrepancies between neutron scattering experiments
and early semiempirical calculations, as well as a recent frozen-magnon
calculation, were resolved [23][see
also: FKF Annual Report 1998, I-57-59]. Savrasov's
LmtARTprogram
is free software which besides FP-LMTO and linear-response includes TB-LMTO-ASA.
A Windows surface is available.
describing dynamical exchange-correlation effects in time-dependent DF
theory, the so-called adiabatic GGA was adopted. The efficiency of the
approach was demonstrated by applications to the transverse spin fluctuations
at T=0 in ferromagnetic Fe and Ni, as well as to the paramagnetic
response in Cr and Pd. Some discrepancies between neutron scattering experiments
and early semiempirical calculations, as well as a recent frozen-magnon
calculation, were resolved [23][see
also: FKF Annual Report 1998, I-57-59]. Savrasov's
LmtARTprogram
is free software which besides FP-LMTO and linear-response includes TB-LMTO-ASA.
A Windows surface is available.
LDA+U calculations of ![]() for CaCuO2 were initiated, but at that stage it seemed more
fruitful that Savrasov go to Rutgers and join the effort initiated by G.
Kotliar, A. Georges, and A. Lichtenstein to develop a method with which
it should be possible to describe strongly correlated, metallic,
real systems. This method is based on dynamical mean-field theory,
which includes the energy- but not the momentum- dependence of the self-energy
through mapping of the Hubbard Hamiltonian -obtained from DF-LMTO calculations-
onto the Anderson impurity model, subject to a selfconsistency condition.
for CaCuO2 were initiated, but at that stage it seemed more
fruitful that Savrasov go to Rutgers and join the effort initiated by G.
Kotliar, A. Georges, and A. Lichtenstein to develop a method with which
it should be possible to describe strongly correlated, metallic,
real systems. This method is based on dynamical mean-field theory,
which includes the energy- but not the momentum- dependence of the self-energy
through mapping of the Hubbard Hamiltonian -obtained from DF-LMTO calculations-
onto the Anderson impurity model, subject to a selfconsistency condition.
We recently succeeded in deriving 3rd-generation MTOs which treat
the energy-dependence in the interstitial region and in the downfolded
channels on the same footing as in the active channels of the spherical
regions, and yet keep the short range [24(a),24(b),
13(a),
13(b)].
The first step is to generate so-called screened spherical waves, ![]() .
These are wave-equation solutions in the interstitial with the following
boundary conditions:
.
These are wave-equation solutions in the interstitial with the following
boundary conditions: ![]() equals
equals ![]() at its own sphere and angular momentum, and it vanishes at any other sphere
or angular momentum, except in the channels to be downfolded which
have logarithmic derivatives equal to
at its own sphere and angular momentum, and it vanishes at any other sphere
or angular momentum, except in the channels to be downfolded which
have logarithmic derivatives equal to ![]() The screened spherical waves are localized as long as their energy is below
the bottom of the hard-sphere continuum. Next, to each solution,
The screened spherical waves are localized as long as their energy is below
the bottom of the hard-sphere continuum. Next, to each solution, ![]() of Schrödinger's equation inside a sphere, we attach the corresponding
screened spherical wave as a tail extending into the interstitial, and
thus form the kinked partial wave
of Schrödinger's equation inside a sphere, we attach the corresponding
screened spherical wave as a tail extending into the interstitial, and
thus form the kinked partial wave ![]() .
The set of kinked partial waves is characterized by a matrix whose element
KR'l'm',Rlm(e)
is the kink of
.
The set of kinked partial waves is characterized by a matrix whose element
KR'l'm',Rlm(e)
is the kink of ![]() in the R'l'm'-channel. This kink matrix is generated from the structure
matrix and the potential parameters by inversion of positive definite matrices
for small clusters. Now, the solutions of Schrödinger's equation at
energy e may be expressed as those linear combinations,
in the R'l'm'-channel. This kink matrix is generated from the structure
matrix and the potential parameters by inversion of positive definite matrices
for small clusters. Now, the solutions of Schrödinger's equation at
energy e may be expressed as those linear combinations, ![]() of kinked partial waves which are smooth, i.e. those for which all kinks
vanish: SRlmKR'l'm',Rlm(e)cRlm,k
=0 for all R'l'm'. Hence, by substituting the partial waves,
which in the ASA are truncated outside the atomic spheres, with kinked
partial waves, we have recovered the ASA formalism without invoking
this approximation.
of kinked partial waves which are smooth, i.e. those for which all kinks
vanish: SRlmKR'l'm',Rlm(e)cRlm,k
=0 for all R'l'm'. Hence, by substituting the partial waves,
which in the ASA are truncated outside the atomic spheres, with kinked
partial waves, we have recovered the ASA formalism without invoking
this approximation.
It turns out that the kinked partial waves span the solutions of Schrödinger's equation for the MT-potential to leading order in the overlap of the spherically symmetric potential wells. As a consequence, the 3rd-generation MTOs are so accurate, that only minimal sets and atom-centered MT-wells are required. This has been confirmed in extensive test calculations for diamond-structured silicon. We find that an sp3-set suffices to describe the valence band and that only when the potential-overlap exceeds 30 per cent in radius, must the overlap correction to the kinetic energy be included explicitly. This correction treats overlaps up to about 60 per cent accurately. Since MT-spheres that large are like van der Waal's spheres, we believe that 3rd-generation MTOs will be useful not only for bulk solids, but also for molecules. We have shown that the overlapping MT-potential should be the least-squares fit to the full potential, weighted with the valence-charge density, and we have sought ways to solve the resulting coupled radial equations efficiently. The total energies calculated from the full charge density have been found to account well for the pressure-volume curves for the crystalline phases of silicon [24(a), 24(b)].
The kink-cancellation equation, ![]() is a screened version of the so-called KKR equation of multiple-scattering
theory. However, rather than energy-dependent kinked partial waves, we
want a minimal set of energy-independent orbitals which span the
solutions of Schrödinger's equation in some range of energies, say
the range of the valence band in Si or the range of the conduction band
in a HTSC. In the spirit of polynomial approximation, we specify a mesh
of N+1 energies, e0,
...,
eN and demand that the
set of Nth-order MTOs (NMTOs) span the solutions of Schrödinger's
equation to within an error proportional to
is a screened version of the so-called KKR equation of multiple-scattering
theory. However, rather than energy-dependent kinked partial waves, we
want a minimal set of energy-independent orbitals which span the
solutions of Schrödinger's equation in some range of energies, say
the range of the valence band in Si or the range of the conduction band
in a HTSC. In the spirit of polynomial approximation, we specify a mesh
of N+1 energies, e0,
...,
eN and demand that the
set of Nth-order MTOs (NMTOs) span the solutions of Schrödinger's
equation to within an error proportional to ![]() .
The set of 0th-order MTOs is obviously the set of kinked partial waves
at the energy e0, and the
corresponding Hamiltonian and overlap matrices are respectively
.
The set of 0th-order MTOs is obviously the set of kinked partial waves
at the energy e0, and the
corresponding Hamiltonian and overlap matrices are respectively ![]() and
and ![]() . For
N=1,
one obtains a set of LMTOs expressed in terms of f(e0,r),
f(e1,r),
K(e0)
and K(e1).
Although it is more practical to use discrete energies, we may let e1
approach e0 and find that
the LMTO becomes
. For
N=1,
one obtains a set of LMTOs expressed in terms of f(e0,r),
f(e1,r),
K(e0)
and K(e1).
Although it is more practical to use discrete energies, we may let e1
approach e0 and find that
the LMTO becomes ![]() This is just like in the ASA-formalism, except that
This is just like in the ASA-formalism, except that ![]() is now a matrix. It turns out that the LMTO Hamiltonian and overlap matrices
are expressed in terms of K(e0),
is now a matrix. It turns out that the LMTO Hamiltonian and overlap matrices
are expressed in terms of K(e0), ![]() ,
K(e1),
and
,
K(e1),
and ![]() . We
have derived and demonstrated the use of a formalism for MTOs of arbitrary
order [13(a), 13(b)].
A summary may be found in the FKF Annual Report
1999 on pp I-16-23. Fig. 1 of this report shows 0th and 1st-order MTOs
for Si and demonstrates the point that, as N increases, each NMTO
becomes more smooth and less localized. This is the way in which the set
can cover a wider energy range without increasing its size. Fig. 3 demonstrates
for GaAs how well the 18 valence and lowest conduction bands over a 35
eV range, including the Ga 3d semi-core bands, are described with
the minimal set of 9 Ga
sp3d5
and 16 As sp3d5f7QMTOs.
Fig. 4 finally demonstrates how well a single orbital, like the one on
the front page, obtained by downfolding of all partial waves except
Cu dx2-y2
can pick out the conduction band of a HTSC.
. We
have derived and demonstrated the use of a formalism for MTOs of arbitrary
order [13(a), 13(b)].
A summary may be found in the FKF Annual Report
1999 on pp I-16-23. Fig. 1 of this report shows 0th and 1st-order MTOs
for Si and demonstrates the point that, as N increases, each NMTO
becomes more smooth and less localized. This is the way in which the set
can cover a wider energy range without increasing its size. Fig. 3 demonstrates
for GaAs how well the 18 valence and lowest conduction bands over a 35
eV range, including the Ga 3d semi-core bands, are described with
the minimal set of 9 Ga
sp3d5
and 16 As sp3d5f7QMTOs.
Fig. 4 finally demonstrates how well a single orbital, like the one on
the front page, obtained by downfolding of all partial waves except
Cu dx2-y2
can pick out the conduction band of a HTSC.
In conclusion, the 3rd-generation formalism solves the long-standing
problem of deriving useful minimal sets of short-ranged orbitals from scattering
theory. Enter into a calculation: 1) The phase shifts of the (overlapping)
potential wells. 2) A choice of which orbitals to include in the set. 3)
For these, a choice of screening radii to control the orbital ranges. 4)
An energy mesh on which the set will provide exact solutions.
References